本文公式与图片来自
Predictions of Hole Mobilities in Oligoacene Organic Semiconductors from Quantum Mechanical Calculations
Wei-Qiao Deng and William A. Goddard
The Journal of Physical Chemistry B 2004 108 (25), 8614-8621
内重组能反映了在电荷转移过程中分子结构形变的程度,而外重组能反映在电荷转移过程中周围分子的弛豫。由于在大多数有机半导体材料中,内重组能是远远大于外重组能,因此我们将忽略外重组能。
在有机半导体分子电荷传输中,初始能量=E(中性分子)+E+(带电荷分子),在电荷跳跃的一瞬间,分子来不及改变结构,因此其末态能量=E*+(中性结构带电荷)+E*(带电荷结构不带电荷)
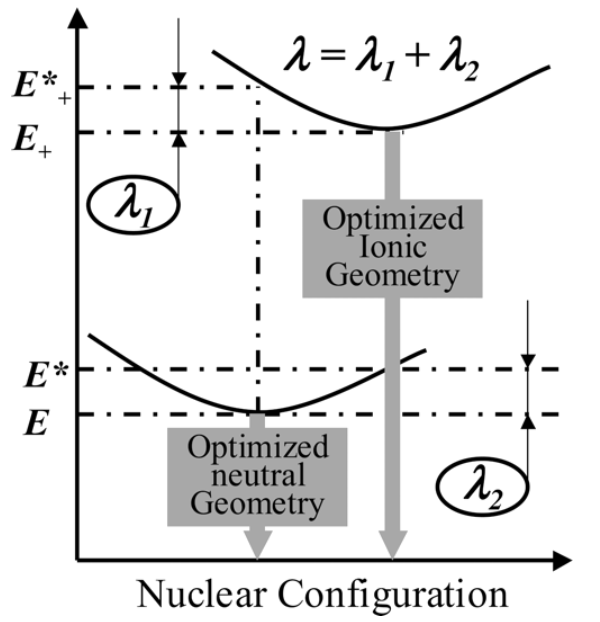
因此重组能计算公式为:
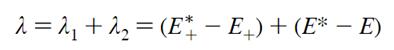
使用Gaussian的计算过程如下:
(1) 优化中性分子结构
(2) 在优化后的中性分子的结构下计算中性分子的能量E
(3) 在优化后的中性分子的结构计算带电荷分子的能量E*+
(4) 优化带电荷分子结构
(5) 在优化后的带电荷分子的结构下计算带电荷分子的能量E+
(6) 在优化后的带电荷分子的结构下计算中性分子的能量E*
其中优化过程的Gaussian输入文件如下
%nprocshared=24 #设定多少核
%mem=48GB #设定内存
%chk=./1.chk #保留chk文件
%rwf=./1.rwf #保留rwf文件
#opt b3lyp/6-311g** #计算的任务类型、泛函、基组
#空行
1 #名字,随便写
#空行
0 1 #电荷与自旋多重度
原子坐标计算能量过程的Gaussian输入文件命令如下
%nprocshared=24
%mem=48GB
%chk=./1.chk
%rwf=./1.rwf
#sp b3lyp/6-311g**计算出所有能量之后,代入公式计算重组能

Comments NOTHING